")
Current Issues
Case Report
Autoimmune lymphoproliferative syndrome presenting as facial panniculitis in a young boy - A case report
*Corresponding Author
Dr. Shailaja Prabhala,
Professor, Department of Pathology,
Kamineni Academy of Medical Sciences & Research centre, LB Nagar, Hyderabad Ranga reddy district, Telangana state, India – 500068.
Email:shailajaprabhala@yahoo.co.in
*1Professor, Department of Pathology, 2Assistant professor, 3Professor and Head, Department of Dermatology and Venereology, Kamineni Academy of Medical Sciences and Research Centre, LB Nagar, Hyderabad, Telangana State, India.
Abstract
A fourteen year old boy presented with gradually enlarging pigmented cutaneous lesions of face and cervical lymphadenopathy for six months. He was evaluated at other hospitals with fine needle aspiration cytology (FNAC) of the parotid gland and later with a guided FNAC of the right external iliac lymph node. He came to our institute due to persistence of the lesions and was evaluated with a skin biopsy and cervical lymph node biopsy. The skin biopsy showed features of panniculitis with emperipolesis. The lymph node revealed prominent emperipolesis and follicular hyperplasia, suggestive of Rosai-Dorfman disease (RDD). Considering the patient’s clinical presentation, previous serological and imaging findings, and the present biopsy reports, a diagnosis of autoimmune lymphoproliferative syndrome was given, and genetic studies for the same were suggested. This case report is presented to draw attention to this rare entity and more so to its unusual presentation as facial panniculitis.
Key Words: Autoimmune lymphoproliferative syndrome, Panniculitis, Rosai-Dorfman disease.
Introduction
Autoimmune lymphoproliferative syndrome (ALPS) is the first disease known to be caused by a primary defect in apoptosis and is the first autoimmune disease with a defined genetic basis.1 It is characterized by nonmalignant lymphadenopathy, splenomegaly and autoimmune cytopenia.2 To meet the case definition of ALPS, a patient must have chronic, nonmalignant lymphadenopathy or splenomegaly of six months or longer duration.3
Case Report
Fig. 1: Clinical photograph at initial presentation
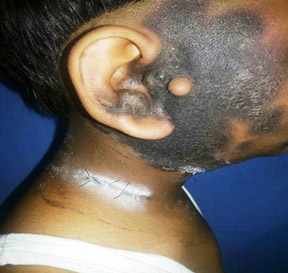 A fourteen year old boy was referred from a private clinic to the Oncology department of our institute for evaluation with a provisional diagnosis of lymphoma. His chief complaints were brownish discoloration of right cheek and nasal bridge which had initially started as a small nodule below the right ear lobule and simultaneous appearance of single left cervical lymphnode (Fig.1). At an outside hospital, FNAC was done from this nodule, and was reported as basal cell adenoma of the parotid gland. Over a six month period, the facial discoloration increased and more cervical lymph nodes appeared. The patient sought treatment at another hospital after two months of initial complaints where a contrast enhanced computed tomography (CECT) of neck showed bilateral parotitis with extensive right cervical lymphadenopathy, possibly of infective origin.
A fourteen year old boy was referred from a private clinic to the Oncology department of our institute for evaluation with a provisional diagnosis of lymphoma. His chief complaints were brownish discoloration of right cheek and nasal bridge which had initially started as a small nodule below the right ear lobule and simultaneous appearance of single left cervical lymphnode (Fig.1). At an outside hospital, FNAC was done from this nodule, and was reported as basal cell adenoma of the parotid gland. Over a six month period, the facial discoloration increased and more cervical lymph nodes appeared. The patient sought treatment at another hospital after two months of initial complaints where a contrast enhanced computed tomography (CECT) of neck showed bilateral parotitis with extensive right cervical lymphadenopathy, possibly of infective origin.
A whole body positron emission tomography (PET) and CECT scans revealed bilateral parotid, submandibular and tonsillar enlargement with hypermetabolic lymph node groups above and below diaphragm, associated with inflammatory component and hypermetabolic hepatosplenomegaly. His antinuclear antibody (ANA) profile was negative. An ultrasound guided FNAC was done at outside hospital from right external iliac lymph node which was reported as reactive in nature with a note that tuberculosis could not be ruled out. There was no past history of tuberculosis, no history of consanguinity in parents, or of any autoimmune diseases.
Cutaneous examination revealed a brownish, hyperpigmented, indurated, non-tender plaque of 12 x 10 cm over the right temple area, extending on to the cheek and retroauricular area. Bilateral cervical lymph nodes were palpable, more on right side 3 x 2.5 cm, firm, non-tender. Rest of the general and systemic examination was normal. Liver and spleen were non-palpable.
The complete blood picture (CBP) was done twice in six months and both the times mild leucopenia was observed. The rest of the parameters of CBP were within normal limits. The erythrocyte sedimentation rate (ESR) was 72mm/1st hour, total serum protein was 8.0gm/dl, serum albumin 3.1gm/dl and globulin was 4.9gm/dl. The rest of the liver function tests were normal. In our institute the CBP showed mild leucopenia and all other parameters were normal. After preoperative investigations, right cervical lymph node biopsy and a deep incision skin biopsy were done as the cutaneous lesions were well established by then.
On histopathological examination, the skin biopsy showed features of septal and lobular panniculitis and evidence of emperipolesis. There was a diffuse lymphohistiocytic infiltrate with collections of histiocytes, some binucleated, in the dermis. There was no evidence of vasculitis or any necrosis. (Fig. 2 and Fig. 3). The lymph nodal masses were 2.5 x 2 x1 cm and 1.5 x 1 x 1 cm. The cut surface of larger mass showed two adherent nodes having thick, inflamed, fibrous capsule, greyish tan surface, and the other showed a single enlarged node with areas of necrosis.

Fig. 2: Skin biopsy showing panniculitis with lymphohistiocytic infiltrate within the subcutaneous adipose tissue. (Hematoxylin and eosin, 100 X)

Fig. 3: Numerous histiocytes with emperipolesis (marked with arrow) in the subcutaneous adipose tissue (Hematoxylin and eosin, 400 X)
Microscopy showed partial effacement of the architecture and pronounced sinusoidal dilatation, filled by lymphocytes, plasma cells, and numerous histiocytes many of which exhibited emperipolesis. Areas of necrosis were present. There was paracortical expansion and also many reactive follicles were present. No fibrosis or parasites were seen (Fig. 4).

Fig. 4: Lymph node showing features of emperipolesis (marked with arrow) (Hematoxylin and eosin, 400 X)
Immunohistochemistry (IHC) was done for CD 1a and CD 68 as Langerhans histiocytosis is an important differential diagnosis. CD 1a was negative and CD 68 was positive as expected in RDD/ALPS.1 A diagnosis of autoimmune lymphoproliferative syndrome was suggested, taking into consideration the clinical presentation, evolution of the lesions, imaging details, persistent cytopenia, skin and lymph node biopsy findings.
Discussion
ALPS is characterized by failure of apoptotic mechanisms leading to the accumulation of autoreactive lymphocytes causing lymphadenopathy and splenomegaly. Such lymphocytes may be potentially oncogenic, thereby, increasing the risk of lymphomas and autoimmune diseases.4 It is inherited mostly in an autosomal dominant fashion, sometimes as an autosomal recessive inheritance or very rarely as a somatic mutation in the FAS or FAS-ligand genes. Hence, family history is important but no such history was available in our patient. Our case presented with features similar to those reported by Joseph5 et al and Kianifar6 et al where both their patients were children with generalized/cervical lymphadenopathy, hepatosplenomegaly and cytopenia. Rosai-Dorfman Disease (RDD) is characterized by massive lymphadenopathy usually affecting cervical nodes (87% cases) and is also associated with extranodal involvement, skin and subcutis being the most commonly affected sites ( 16% cases).7 The typical histological finding is emperipolesis where lymphocytes are seen within the histiocytes. RDD has leukocytosis, fever, elevated ESR and polyclonal hypergammaglobulinemia.8 It is worth noting that hepatosplenomegaly is rare in RDD.9 As our patient had persistent cytopenia and hepatosplenomegaly along with features of paracortical expansion and reactive follicles in the lymph node, a diagnosis of ALPS was preferred. As many as 41% patients of type 1 ALPS (FAS gene associated) show RDD-like changes in the lymph nodes suggesting that RDD and ALPS may be related.10 Genetic tests for FAS, FAS-ligand, caspase genes and test to demonstrate double negative lymphocytes in peripheral blood are confirmatory and hence were advised to the patient, but could not be done due to financial constraints.

Fig. 5: Clinical photograph with improvement in skin lesion after two months of treatment
Conclusion
ALPS though a rare entity, should be suspected especially in children who present with persistent or progressive lymphadenopathy and it can present as panniculitis. A high index of suspicion is required so as to avoid unnecessary diagnostic and therapeutic interventions.
References
- John MJ, Rajasekhar R, Mathews V. Autoimmune lymphoproliferative syndrome (ALPS): a rare cause of immune cytopenia. Indian Pediatr.2008;45(2):148-50.
- Canale VC, Smith CH. Chronic lymphadenopathy simulating malignant lymphoma. J Pediatr. 1967;70(6):891-9.
- Fleisher TA. The autoimmune lymphoproliferative syndrome: an experiment of nature involving lymphocyte apoptosis. Immunol Res. 2008;40(1):87-92.
- Rao VK, Straus SE. Causes and consequences of the autoimmune lymphoproliferative syndrome. Hematology. 2006;11(1):15-23.
- Joseph J, Reena R, Vikram M. Autoimmune lymphoproliferative syndrome (ALPS): A rare cause of immune cytopenia. Indian Pediatrics 2008;45:148-50.
- Kianifar MR, Kalesi M, Farid R, Badiee Z, Rastin M, Ahanchian H. Iran J Allergy Asthma Immunol 2010;9(3):181-83.
- Histiocytosis Association, a rare community. Available from: www.histio.org
- Foucar E, Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Review of the entity. Semin Diagn Pathol 1990;7:19-73.
- McClain KL, Natkunam Y, Swerdlow SH. Atypical cellular disorders. Hematology Am Soc Hematol educ Program 2004;283-96.
- Maric I, Pittaluga S, Dale JK, Niemela JE, Delsol G, Diment J, et al. Histologic features of sinus histiocytosis with massive lymphadenopathy in patients with autoimmune lymphoproliferative syndrome. Am J Surg Pathol 2005,29:903-11.