")
Current Issues
Case Report
Epidermolysis Bullosa Acquisita: A rare immune-bullous disorder- A case report
*Corresponding author
Dr Arun Kumar M
Department of Dermatology and Venereology,
Kamineni Institute of Medical Sciences, Narketpally, Nalgonda District, Telangana State, India - 508 254,
Email: drmettaarun@yahoo.co.in
1Professor & Head, 2Assistant professor, 3Post Graduate, Department of Dermatology and Venereology, Kamineni Institute of Medical Sciences, Narketpally, Nalgonda District, Telangana State, India.
ABSTRACT
Epidermolysis bullosa acquisita (EBA) is a chronic mucocutaneous autoimmune blistering disease characterised by skin fragility, blistering at the sites of trauma resulting in scarring and formation of milia. A 21 years old female patient presented with complaints of tense fluid filled lesions on and off since 3 years. There was a history of trauma provoking the onset of lesions and lesions always healed with scarring. There was no family history of similar complaints. She was diagnosed as EBA based on clinical examination, histopathology and immunofloroscence study. Patient was treated with oral prednisolone and dapsone, responded well and was in remission. We are presenting this as an interesting rare case.
Key words :Direct immunofluoroscence, epidermolysis bullosa acquisita, scarring, trauma.
Introduction
Epidermolysis Bullosa (EB) generally refers to a group of inherited disorders that involve the formation of blisters following trivial trauma. Epidermolysis Bullosa Acquisita (EBA) is a chronic subepidermal blistering disease associated with autoimmunity to type-VII collagen within anchoring fibrils located at the dermal-epidermal junction. Although an acquired disease, it was placed in the category of EB 100 years ago, as physicians were struck by its similarity with hereditary epidermolysis bullosa dystrophica. 1 It is a rare, autoimmune sub-epidermal bullous disease of unknown aetiology. It is characterised by skin fragility, blistering at sites of trauma, resulting in scarring and formation of milia. Mucous membrane involvement is variable. Treatment options are limited and often difficult.2
Case Report
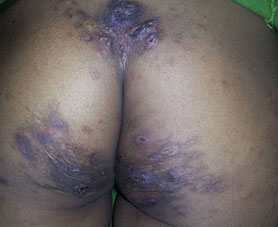
A 21 years old female patient came to the outpatient department of dermatology with asymptomatic fluid filled lesions over the back and extremities on and off since 3 years. Blisters appeared initially over the lower back and similar lesions appeared over elbows and knees within a span of 6 months. These blisters ruptured spontaneously within 4-5 days leaving raw areas which were covered with blood and pus, later dried up to form thick crusts. These lesions healed in about 2 weeks time leaving behind hyperpigmentation and scarring. There was a history of minor injury (pressure/friction/rubbing) prior to the onset of lesions. There was no history of itching or wheals, photosensitivity prior to the onset of lesions. There was no history suggestive of connective tissue disorder. There was no history of erosions involving oral cavity and other mucosal surfaces. There was no history of high coloured urine, irregular bowel habit or relation with any diet. There was no history of intake of systemic drugs and application of topical irritants prior to the onset of lesions. Cutaneous examination revealed multiple, polysized, tense and few flaccid vesiculo-bullous lesions on an erythematous base along with crusted plaques, distributed bilaterally and symmetrically over the sacral area and lower half of buttocks, lateral aspects of thighs, elbows and knees with areas of scarring and hyperpigmentation (Fig.1, Fig.2). Nikolsky’s sign and bulla spread sign were negative. Mucous membranes, hair and nails were normal. Systemic examination was unremarkable.
 Routine haemogram and other biochemical investigations were within normal limits. Histopathology sections showed intact epidermis of normal thickness and a large bullous lesion in the sub epidermal junction, extending up to mid-dermis. It was filled with numerous degenerating and viable neutrophils. Deep dermis and subcutis appeared normal (Fig. 3).
Routine haemogram and other biochemical investigations were within normal limits. Histopathology sections showed intact epidermis of normal thickness and a large bullous lesion in the sub epidermal junction, extending up to mid-dermis. It was filled with numerous degenerating and viable neutrophils. Deep dermis and subcutis appeared normal (Fig. 3).
Differential diagnosis such as cystic neoplasm of pancreas, On direct immunofluorescence (IF), IgG was positive (++) in the bullous lesion, with accentuation of basement membrane zone and IgA, IgM & C3 were negative (Fig 4).


Based on the history, cutaneous examination, histopathology and immunofluorescence study, a diagnosis of epidermolysis bullosa acquisita was made. Patient was started on oral prednisolone (1mg/kg/day) and dapsone (100mg/day) along with topical clobetesol propionate (0.05%) with sodium fusidate cream as local application. Patient responded very well to the medication and is on regular follow up till date.
Discussion
Epidermolysis bullosa acquisita (EBA) is a chronic mucocutaneous autoimmune blistering disease. The pathogenic relevance of autoantibodies targeting type VII collagen (COL7) has been well-documented. Criteria of Roenigk3,4 to diagnose EBA include (i) clinical lesions resembling epidermolysis bullosa dystrophica, (ii) adult onset of disease, (iii) a negative family history of epidermolysis bullosa dystrophica, and (iv) exclusion of other bullous diseases.2, 3
The cutaneous manifestations in EBA patients are heterogeneous. However, EBA patients can be classified into two major clinical subtypes: non-inflammatory (classical or mechanobullous) and inflammatory EBA, which is characterized by cutaneous inflammation resembling bullous pemphigoid, linear IgA disease, mucous membrane pemphigoid, or Brunsting-Perry pemphigoid.4-6 Blisters may be serous or haemorrhagic, tend to be localized to areas of trauma (especially the dorsal aspect of the hands and feet, and elbows), and heal with scarring, milia formation, and hyperpigmentation. The nails may be dystrophic but are often normal. Mucosal involvement is variable.7 Extra-cutaneous EBA manifestations include ocular, oral mucosa, oesophagus, anal, vaginal, tracheal, and laryngeal lesions. Ocular involvement in EBA predominantly presents with scarring, resembling the lesions observed in patients with mucous membrane pemphigoid.4,5,8
The diagnosis of EBA is based on the clinical presentation, the detection of tissue-bound antibodies by direct immunofluorescence (DIF) microscopy, and the detection of circulating antibodies directed against COL7 and/or a u-serrated pattern in direct IF microscopy. The localization of immune deposits within the dermal-epidermal junction of the skin of EBA patients by immunoelectron microscopy is considered the gold standard for the diagnosis.1 Other diagnostic investigations are histopathology where subepidermal blister and a clean separation between the epidermis and dermis is seen. Direct and indirect immunofluorescence show dermo-epidermal accentuation similar to bullous pemphigoid. These two conditions can be differentiated by performing salt split immunofluorescence in 1 molar NaCl. Western blot technique and ELISA can also be performed.1, 9
EBA is a very difficult disease to treat. Supportive therapy, such as instructions in wound care and strategies for avoiding all sorts of trauma, is warranted in all patients of EBA. The patients must be educated to recognize skin infections and to seek medical care and antibiotic therapy promptly. Systemic glucocorticoids, azathioprine, methotrexate, and cyclophosphamide are mostly ineffective, even in large doses. Some patients improve with dapsone, especially when neutrophils are present in their dermal infiltrate. Cyclosporine is often effective, but has long-term toxicity. Colchicine in high doses is helpful in many cases, provided the patient can tolerate its gastrointestinal side effects.1
Complications of EBA include secondary skin infections, usually due to Staphylococcus and Streptococcus, because the blisters and erosions compromise the skin’s barrier. Severe EBA patients may develop significant fibrosis of the hands with decreased range of motion of the palms and digits. EBA patients with significant mucosal involvement may develop oesophageal strictures and even laryngeal scarring.9
There is an association with inflammatory bowel disease both ulcerative colitis and Crohn’s disease, multiple myeloma, amyloidosis, lymphoma and systemic lupus erythematosus. Drug induced EBA has also been reported.8,9
Conclusion
Clinical relevance of categorising Epidermolysis Bullosa Acquisita into various subtypes is important for any treating physician from prognostic perspective. As the current case falls under inflammatory clinical subtype, it responded well to the anti-inflammatory agents.
References:
- Das JK, Sengupta S, Gangopadhyay AK. Epidermolysis bullosa acquisita. Indian J Dermatol Venereol Leprol 2006;72:86.
- Nair VL, Ganga P. Epidermolysis bullosa acquisita. Indian J Dermatol Venereol Leprol 1990;56:310-11.
- Gangopadhyay AK. Epidermolysis bullosa acquisita. Indian J Dermatol 1997;42:121-22.
- Ishii N, Hamada T, Dainichi T, Karashima T, Nakama T, Yasumoto S, et al., Epidermolysis bullosa acquisita: What's new? J Dermatol. 2010;37:220-30.
- Schmidt E D. Pemphigoid diseases. The Lancet 2013;381:320-32.
- Wojnarowska F, Eady RA, Burge SM. Bullous eruptions. In: Champion RDH, Burton JL, Burn DA, Breathnach SM, editors. Rook’s Textbook of Dermatology. 6th ed. Oxford: Blackwell Science, 1998; 1817-97.
- Chen M, Prakash L, Woodley DT. Epidermolysis bullosa acquisita: Autoimmunity to anchoring fibril collagen. Autoimmunity 2012;45:91-101.
- Wojnarowska F, Venning VA. Immunobullous Diseases. In: Burns T, Breathnach S, Cox N, Griffiths C, editors. Rook’s Textbook of Dermatology. 8th ed. Oxford: Blackwell Science; 2010. 40. p. 51-55.